
特別說明:本文由學(xué)研匯技術(shù) 中心原創(chuàng)撰寫,旨在分享相關(guān)科研知識。因?qū)W識有限,難免有所疏漏和錯(cuò)誤,請讀者批判性閱讀,也懇請大方之家批評指正。
原創(chuàng)丨彤心未泯(學(xué)研匯 技術(shù)中心)
研究背景
有機(jī)分子中C-H鍵的官能化是化學(xué)合成最直接的方法之一。定向C-H活化可以通過避免冗長的預(yù)活化和功能化序列來改變有機(jī)分子的合成,被認(rèn)為極具潛力的研究方向。隨著催化的不斷發(fā)展,目前已經(jīng)實(shí)現(xiàn)了羧酸、酮和胺等天然化學(xué)基團(tuán)控制和指導(dǎo)C(sp3)–H 活化。
關(guān)鍵問題
1、醇作為最常見天然官能團(tuán),仍未實(shí)現(xiàn)C-H的定向活化醇作為有機(jī)化學(xué)中最常見的官能團(tuán)之一,由于其對后過渡金屬催化劑的親和力較低,在C-H活化反應(yīng)中仍存在極大局限性。2、醇作為典型的導(dǎo)向基團(tuán),反應(yīng)僅局限于簡單的C(sp2)-H鍵斷裂盡管醇是經(jīng)典有機(jī)反應(yīng)(包括環(huán)氧化、環(huán)丙烷化、氫化和加氫金屬化)中的典型導(dǎo)向基團(tuán),但很少有游離醇導(dǎo)向的C-H活化反應(yīng)被報(bào)道,且所有反應(yīng)都僅限于更容易的C(sp2 )-H鍵斷裂。3、醇衍生物導(dǎo)向C(sp3)-H活化的實(shí)用性受限到目前為止,報(bào)道的正式羥基定向C(sp3)–H活化方法都依賴于醇的衍生化來構(gòu)建更強(qiáng)的配位定向基團(tuán),但它們的實(shí)用性往往受到合成、構(gòu)建和去除導(dǎo)向基團(tuán)的必要性的限制。
新思路
有鑒于此,美國斯克里普斯研究所余金權(quán)等人利用電荷平衡和氫鍵相互作用實(shí)現(xiàn)了L 型羥基與鈀的穩(wěn)定配位,進(jìn)而促進(jìn)關(guān)鍵C-H裂解轉(zhuǎn)變的組裝狀態(tài),最終實(shí)現(xiàn)了δ-C(sp3)–H 鍵的醇定向芳基化配體。在研究過程中,電荷平衡和二級配位層氫鍵相互作用已通過結(jié)構(gòu)-活性關(guān)系研究、計(jì)算模型和晶體學(xué)數(shù)據(jù)等多種數(shù)據(jù)證明。之前在C-H活化中的研究表明在既定的反應(yīng)性背景下,二次相互作用被用于控制選擇性,而本報(bào)告證明了利用二次相互作用通過增強(qiáng)底物-催化劑親和力來實(shí)現(xiàn)具有挑戰(zhàn)性的、以前未知的反應(yīng)性的可行性。作者提出選擇性加強(qiáng)固化羥基與Pd(II)配位的配體可以實(shí)現(xiàn)游離醇引導(dǎo)的C(sp3)–H活化,并通過開發(fā)兩類不同底物的游離醇證實(shí)了這一策略的可行性。2、構(gòu)建了模型系統(tǒng)并進(jìn)行了配體優(yōu)化作者選擇苯甲醇1a作為模型底物確定了一個(gè)簡單的模型系統(tǒng),證實(shí)了雙陰離子配體對羥基導(dǎo)向基團(tuán)的L型配位的重要性。3、環(huán)丁烷醇誘導(dǎo)δ-芳基化配體優(yōu)化和范圍作者進(jìn)一步將該策略擴(kuò)展到其他底物,證實(shí)了該策略在環(huán)丁醇、苯乙基衍生物、芳基環(huán)戊烷等同樣有效,表明反應(yīng)活性并不限于環(huán)丁基C-H鍵。作者通過X射線衍射和晶體結(jié)構(gòu)證實(shí)了醇導(dǎo)向基團(tuán)與配體之間的氫鍵相互作用,通過理論計(jì)算表明氫鍵在實(shí)現(xiàn)醇引導(dǎo)的C(sp3)–H活化中的重要作用。1、提出了實(shí)現(xiàn)游離醇誘導(dǎo)C(sp3)-H活化的可行性策略作者提出,可以選擇性加強(qiáng)固定羥基與Pd(II)配位的配體實(shí)現(xiàn)游離醇誘導(dǎo)的C(sp3)–H活化,該策略依賴于庫侖電荷平衡有利于中性醇導(dǎo)向基團(tuán)的配位及納入二級配位球氫鍵相互作用以穩(wěn)定底物-催化劑絡(luò)合。2、開發(fā)了兩類不同底物的游離醇導(dǎo)向的δ-C(sp3)–H芳基化作者通過開發(fā)兩類不同底物的游離醇導(dǎo)向的δ-C(sp3)–H芳基化,證實(shí)了上述策略的可行性。作者提出,選擇性加強(qiáng)固化羥基與Pd(II)配位的配體可以實(shí)現(xiàn)游離醇引導(dǎo)的 C(sp3)–H活化。該策略依賴于兩個(gè)關(guān)鍵原則:庫侖電荷平衡有利于中性醇導(dǎo)向基團(tuán)的配位,及納入二級配位球氫鍵相互作用以穩(wěn)定底物-催化劑絡(luò)合,并通過限制Pd-O鍵旋轉(zhuǎn)來幫助組織CMD過渡態(tài)。通過開發(fā)兩類不同底物的游離醇導(dǎo)向的δ-C(sp3)–H芳基化,證實(shí)了這一策略的可行性。
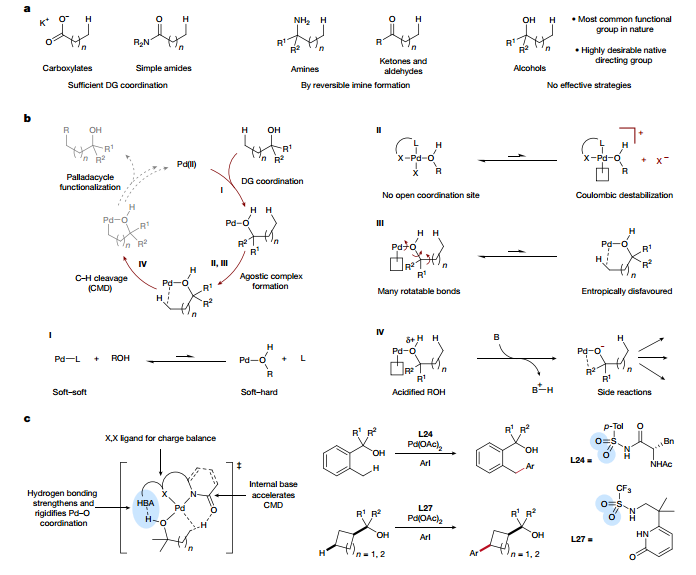
圖 實(shí)現(xiàn)游離醇定向C(sp3)–H激活的價(jià)值、挑戰(zhàn)和策略構(gòu)建模型系統(tǒng)并進(jìn)行配體優(yōu)化為了探索配體結(jié)構(gòu)-活性關(guān)系(SAR),作者確定了一個(gè)簡單的模型系統(tǒng)。作者提出游離醇導(dǎo)向的反應(yīng)性更適合參與遠(yuǎn)程位置,選擇苯甲醇1a作為模型底物,發(fā)現(xiàn)它在Pd(OAc)2存在下發(fā)生可測量的C(sp3)-H芳基化。在最佳配體存在下進(jìn)行的可比添加劑研究以及催化劑SAR數(shù)據(jù)、晶體學(xué)證據(jù)和計(jì)算模型,解析了醇配位為本研究中描述的反應(yīng)中的中性導(dǎo)向基團(tuán)。此外,作者還證實(shí)了雙陰離子配體對羥基導(dǎo)向基團(tuán)的L型配位的重要性,通過常規(guī)篩選最終確定苯丙氨酸衍生物L24為最佳配體。
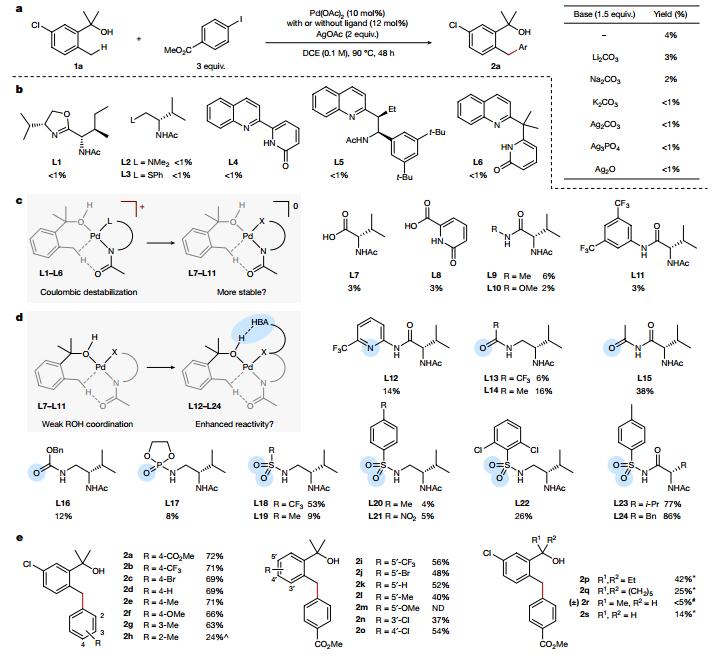
在確定了苯甲基C-H活化反應(yīng)的配體后,作者進(jìn)一步探究了該策略是否可以擴(kuò)展到更具挑戰(zhàn)性的底物。作者探究了環(huán)丁醇的δ-官能化,發(fā)現(xiàn)L24對δ位的芳基化具有獨(dú)特的選擇性,需要增加連接體的靈活性來補(bǔ)償吡啶酮部分的剛性。反應(yīng)范圍的檢查表明,該反應(yīng)對規(guī)模不敏感,多取代和雜環(huán)芳基碘化物也能以中等至高產(chǎn)率進(jìn)行反應(yīng),且該策略對苯乙基衍生物依然有效。芳基環(huán)戊烷同樣可以以適度的產(chǎn)率形成,表明反應(yīng)活性并不限于環(huán)丁基C-H鍵。
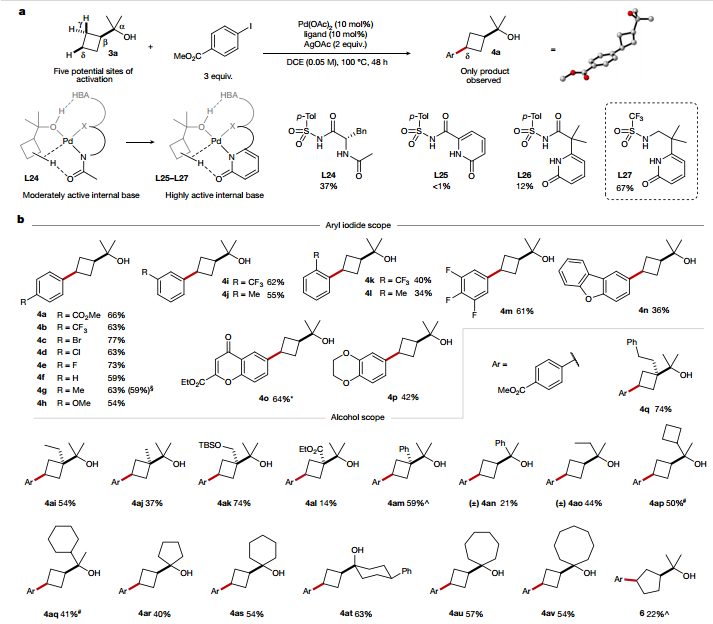
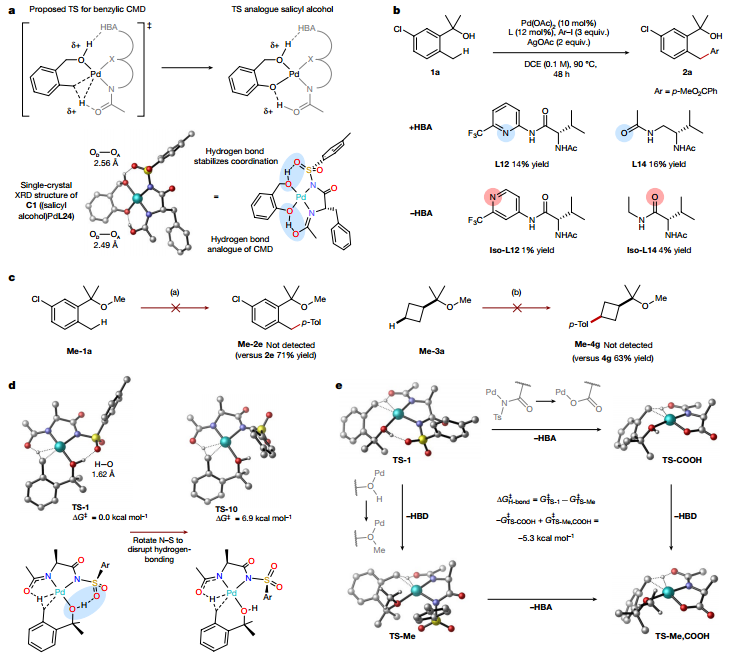
圖 環(huán)丁烷醇誘導(dǎo)δ-芳基化的配體優(yōu)化和范圍最后,作者探究了氫鍵相互作用的有效性。通過X射線衍射證實(shí)了CMD過程中的質(zhì)子轉(zhuǎn)移,并通過晶體結(jié)構(gòu)指出了醇導(dǎo)向基團(tuán)與配體之間的氫鍵相互作用。接著作者探究了氫鍵相互作用對反應(yīng)性的影響,使用DFT模型來研究所提出的氫鍵相互作用對CMD過渡態(tài)的影響,結(jié)果強(qiáng)烈表明氫鍵相互作用為TS-1提供了顯著的穩(wěn)定性。因此,氫鍵在L24-Pd和L27-Pd配合物實(shí)現(xiàn)醇引導(dǎo)的C(sp3)–H活化方面發(fā)揮著關(guān)鍵作用。
展望
總之,作者提出,本工作所展示的策略是解決游離醇定向C(sp3)–H活化的有效方案,特別適合涉及弱協(xié)調(diào)定向基團(tuán)的轉(zhuǎn)化。在熱力學(xué)上不利于底物與催化劑配位的反應(yīng)中,穩(wěn)定底物-催化劑絡(luò)合的非共價(jià)相互作用可能會選擇性地穩(wěn)定整個(gè)催化循環(huán)中涉及催化劑-底物絡(luò)合物的過渡態(tài),從而為該反應(yīng)提供了重要基礎(chǔ)。此外,配體的設(shè)計(jì)將加速關(guān)鍵的基本步驟,進(jìn)而促進(jìn)反應(yīng)的進(jìn)一步發(fā)生。作者認(rèn)為,該策略將廣泛適用于實(shí)現(xiàn)各種具有挑戰(zhàn)性的底物導(dǎo)向的有機(jī)金屬轉(zhuǎn)化。Strassfeld, D.A., Chen, CY., Park, H.S. et al. Hydrogen-bond-acceptor ligands enable distal C(sp3)–H arylation of free alcohols. Nature (2023). https://doi.org/10.1038/s41586-023-06485-8










