
單原子合金最近成為高活性和高選擇性的合金催化劑。與純金屬不同,單原子合金擺脫了近三十年前為預測催化性能而開發的成熟概念框架。盡管這為探索迄今為止無法實現的化學提供了機會,但這讓我們沒有一個簡單的指南來設計能夠催化目標反應的單原子合金。
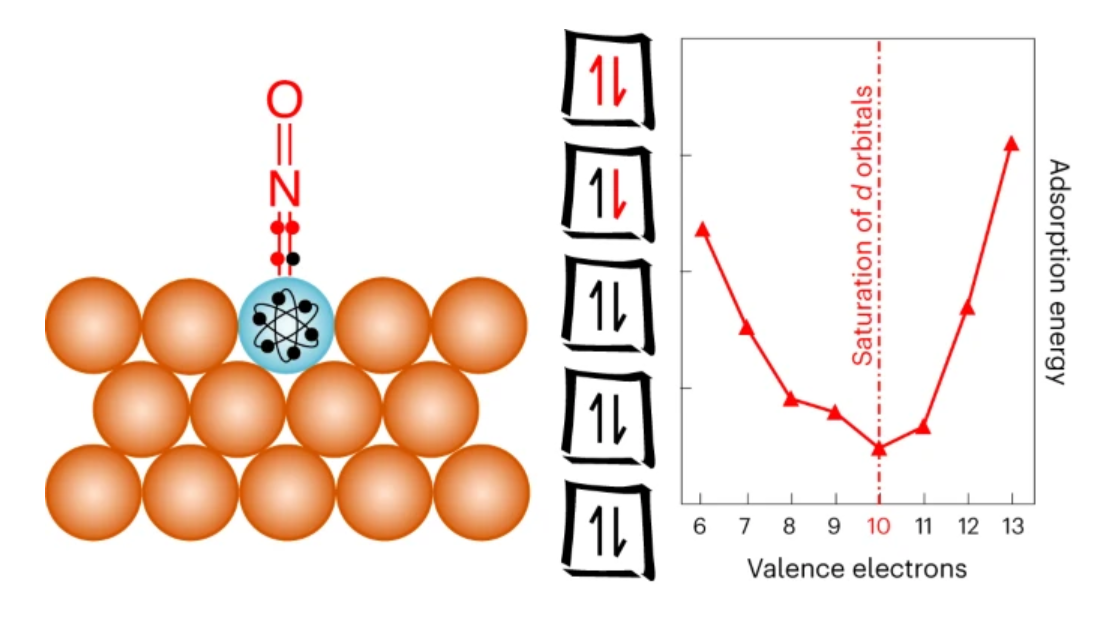
基于數千次密度泛函理論計算,倫敦大學學院Romain Réocreux等揭示了單原子合金表面摻雜劑原子(通常是活性位點)上吸附物結合的10電子計數規則。
本文要點:
(1)
一個簡單的分子軌道方法使這一規則和吸附質-摻雜劑相互作用的性質合理化。此外,作者的直觀模型可以加速單原子合金催化劑的合理設計。事實上,作者說明了電子計數規則提供的獨特見解如何幫助確定工業相關氫化反應中最有前途的摻雜劑,從而將潛在材料的數量減少一個數量級以上。
(2)
該規則為實驗者和理論家提供了有見地的指導,用于設計具有工業意義的目標反應的單原子合金催化劑。
參考文獻:
Schumann, J., Stamatakis, M., Michaelides, A. et al. Ten-electron count rule for the binding of adsorbates on single-atom alloy catalysts. Nat. Chem. (2024).
DOI: 10.1038/s41557-023-01424-6
https://doi.org/10.1038/s41557-023-01424-6

















