
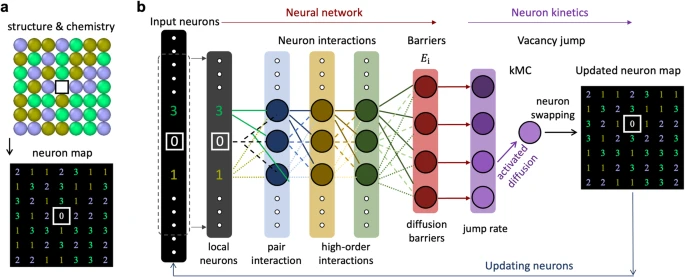
涉及到原子空間傳輸的擴散決定了許多重要的過程和行為,如沉淀和相形核。成分復雜的材料中固有的化學復雜性對原子擴散建模和由此形成的化學有序結構帶來了一定挑戰。鑒于此,來自加州大學的Penghui Cao等人開發了一種神經網絡動力學(NNK)方法,該方法能夠預測和模擬復雜濃縮化學環境中擴散誘導的化學和結構演變。
文章要點:
1) 該研究開發的這種框架是基于高效的晶格結構和化學表示,結合人工神經網絡,能夠精確預測所有路徑相關的遷移勢壘和單個原子跳躍,為了證明該方法的可行性,他們研究了耐火NbMoTa合金中與溫度相關的局部化學有序,并揭示了B2有序達到最大值的臨界溫度;
2) 此外,研究發現,原子跳躍隨機性圖在該特征溫度附近表現出最高的擴散不均勻性(多重性),這與化學有序和B2結構的形成密切相關,可擴展的NNK框架為探索具備潛在優越性質的組成空間中的擴散行為提供了一條可行途徑。
參考資料:
Wang, Y., Zhou, Y., Yang, Q. et al. Self-assembly of nanocrystal checkerboard patterns via non-specific interactions. Nat Commun 15, 3913 (2024).
10.1038/s41467-024-47572-2
https://doi.org/10.1038/s41467-024-47572-2

















